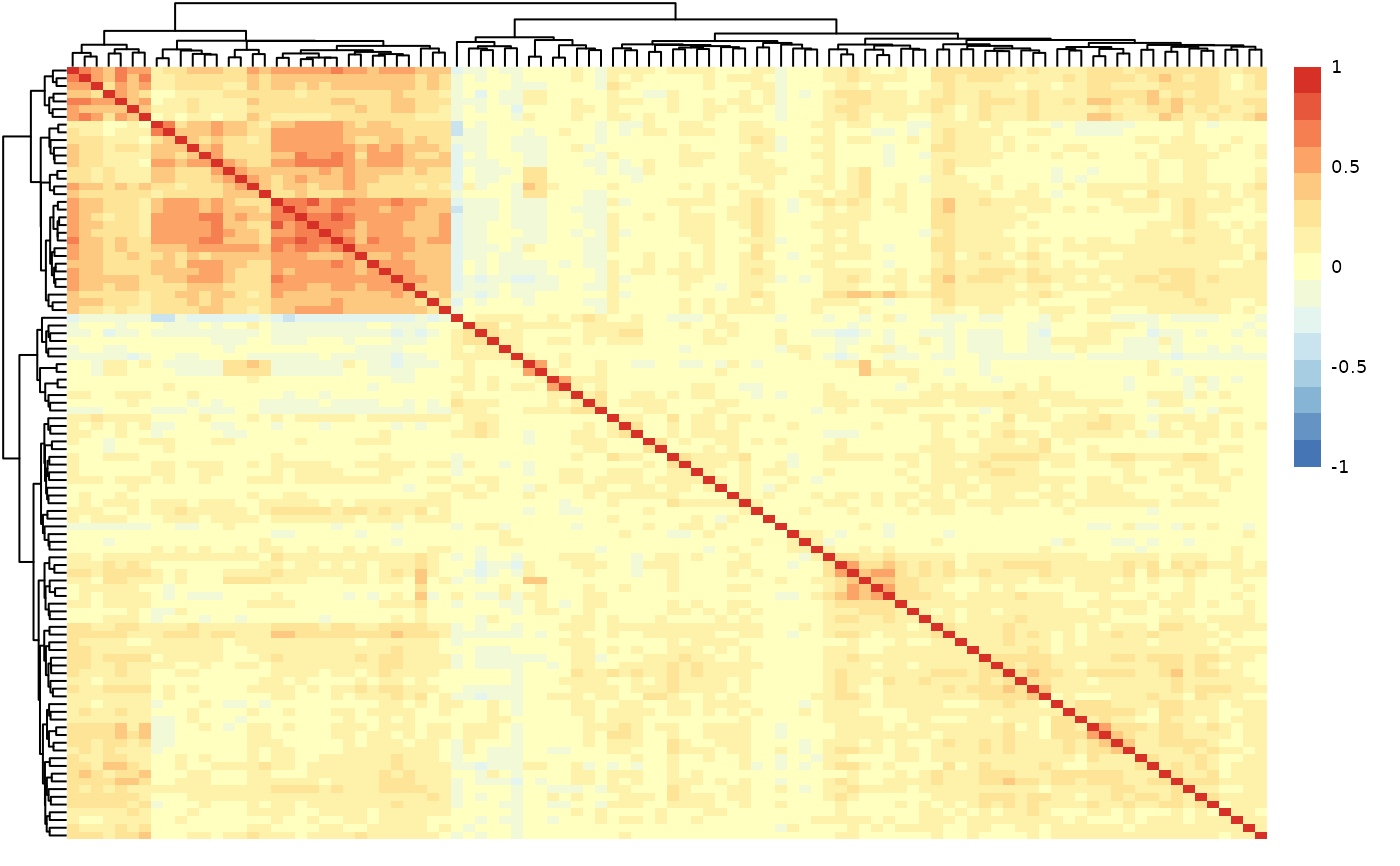
hd_plot_cor_heatmap() calculates the correlation matrix of the input dataset.
It creates a heatmap of the correlation matrix. This matrix is created via hd_correlate().
It also filters the feature pairs with correlation values above the threshold and
returns them in a tibble.
Usage
hd_plot_cor_heatmap(
x,
y = NULL,
use = "pairwise.complete.obs",
method = "pearson",
threshold = 0.8,
cluster_rows = TRUE,
cluster_cols = TRUE
)Arguments
- x
A numeric vector, matrix or data frame.
- y
A numeric vector, matrix or data frame with compatible dimensions with
x. Default is NULL.- use
A character string. The method to use for computing correlations. Default is "pairwise.complete.obs". Other options are "everything", "all.obs", " complete.obs", or "na.or.complete".
- method
A character string. The correlation method to use. Default is "pearson". Other options are "kendall" or "spearman".
- threshold
The reporting correlation threshold. Default is 0.8.
- cluster_rows
Whether to cluster the rows. Default is TRUE.
- cluster_cols
Whether to cluster the columns. Default is TRUE.
Value
A list with the correlation matrix, the filtered pairs and their correlation values, and the heatmap.
Examples
# Prepare data
dat <- example_data |>
dplyr::select(DAid, Assay, NPX) |>
tidyr::pivot_wider(names_from = "Assay", values_from = "NPX") |>
dplyr::select(-DAid)
# Correlate proteins
results <- hd_plot_cor_heatmap(dat, threshold = 0.7)
# Print results
results$cor_matrix[1:5, 1:5] # Subset of the correlation matrix
#> AARSD1 ABL1 ACAA1 ACAN ACE2
#> AARSD1 1.00 0.47 0.19 -0.06 0.04
#> ABL1 0.47 1.00 0.46 -0.01 0.13
#> ACAA1 0.19 0.46 1.00 0.03 0.32
#> ACAN -0.06 -0.01 0.03 1.00 0.07
#> ACE2 0.04 0.13 0.32 0.07 1.00
results$cor_results # Filtered protein pairs exceeding correlation threshold
#> Protein1 Protein2 Correlation
#> 1 ATP5IF1 AIFM1 0.76
#> 2 AXIN1 ARHGEF12 0.76
#> 3 AIFM1 ATP5IF1 0.76
#> 4 ARHGEF12 AXIN1 0.76
#> 5 ARHGEF12 AIFM1 0.71
#> 6 AIFM1 ARHGEF12 0.71
results$cor_heatmap # Heatmap of protein-protein correlations